In the broad areas of statistics, data science and bioinformatics, the central theme of our research is to formulate the data science challenges motivated by biomedical and biotechnological data into computational problems and tackle them by developing novel statistical and AI methodologies as well as analytical workflows to enable new scientific discoveries in various diseases. Our current research focus on the rapidly evolving field of biomedical data science through the following key areas:
AI & machine learning solutions for complex biomedical single-cell and spatial omics data
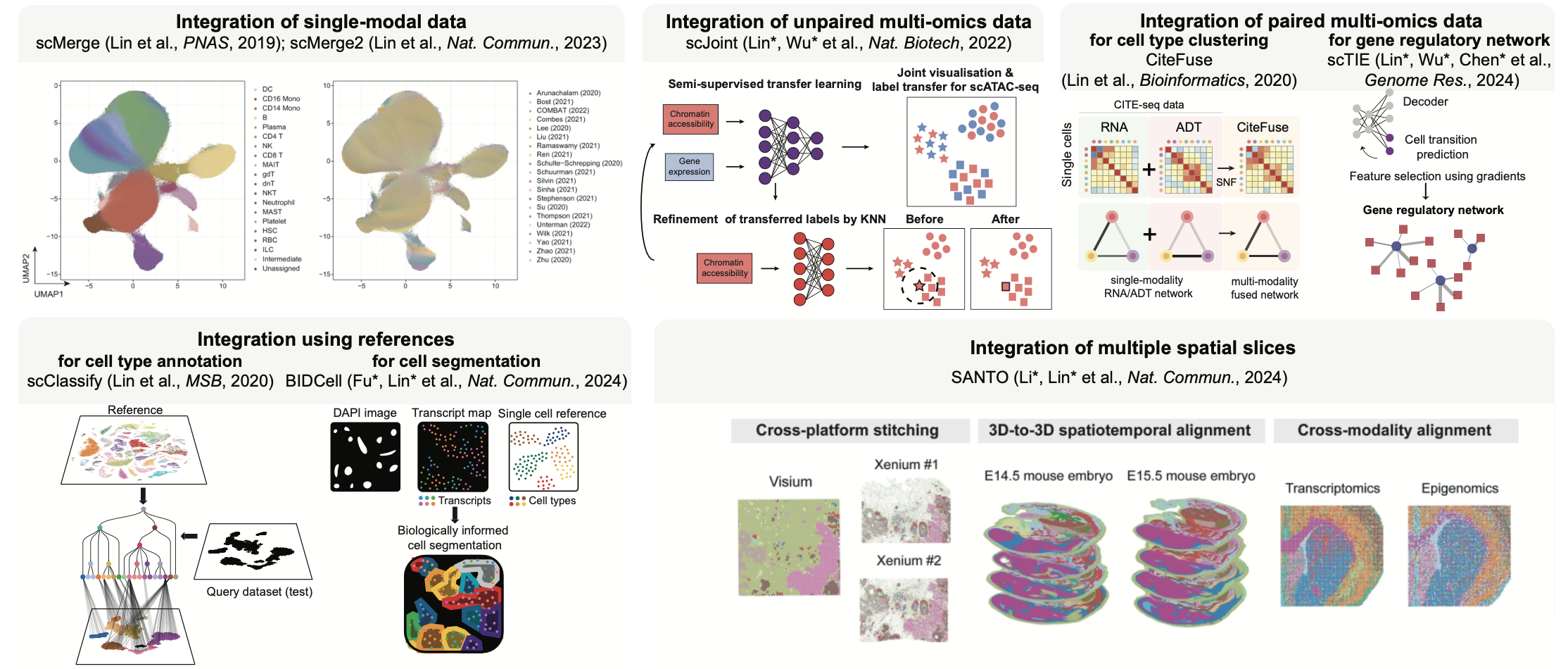
Our research focuses on developing scalable, interpretable and biologically informed AI/machine learning solutions to address these open challenges in data science and translate advanced biomedical data analytics into knowledge and practice, with a focus on:
- Single-cell single-modal data integration
- Single-cell multi-modal data integration
- Spatial transcriptomics alignment
- Spatial multi-modal data integration across time points
Interdisciplinary collaboration and creative data analytics in biomedical research
Our work is done in collaboration with researchers in researchers from various disciplines, including neuroscience, cancer biology, transplantation and biotechnology. Many of our research contribute to novel analytics in collaborative papers. These papers highlight how innovative statistical methodologies and data analytics contribute to biomedical research for various diseases and biological processes, including tendon repair (Chen et al., 2025), melanoma (Lim et al., 2024), brain development (Zhang et al., 2025), PTSD (Hwang et al., 2025), COVID-19 (Lin et al., 2022) and kidney transplant (Lin et al., 2023, Lin et al., 2022).
